Find interstitial positions / sites in crystal structure automatically using the Voronoi or charge density method, and create a modified structures with specified number of interstitials that are farthest, closest, or moderately spaced from one another
You can also use the following online application to create supercells and generate interstitials, vacancies, or substitutes: 🔬 XRDlicious point defects generator
Articles to cite when using pymatgen and pymatgen.analysis.defects modules:
Prerequisities
- pymatgen
- pymatgen.analysis.defects
- Python 3.x
How to use the code
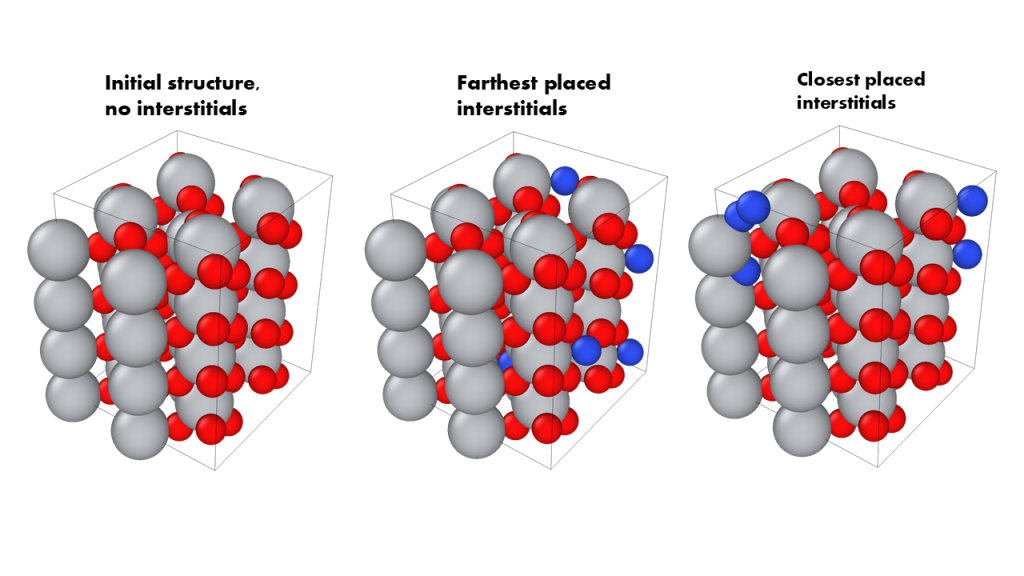
Download the python script ‚calc_int_sites.py‚. It finds the interstitial positions in a crystal structure (in POSCAR format) using the VoronoiInterstitialGenerator from the pymatgen.analysis.defects module. It then selects a specified number of interstitial sites that are maximally, minimally, and moderately (between maximally and minimally) spaced apart and places a chosen interstitial element at these positions. The modified structure with these interstitials are then saved in CIF and POSCAR formats.
Into the same folder where is the calc_int_sites.py script, put also your structure in POSCAR format. If you want to use ChargeInterstitialGenerator method, also put here CHGCAR file corresponding to the POSCAR file.
In the code, change the following user’s settings as needed:
interstitial_element_to_place = "N" # Which element to insert as interstitial
number_of_interstitials_to_insert = 5 # How many elements to insert as interstitials
which_interstitial_to_use = 0 # The value '0' will consider all found available interstitial positions for calculating
#the distances. If you want to place interstitials on only the certain type of interstitial site, e.g., this method will
#identify two different types of interstitial positions and you want to insert interstitial only on the first type,
#then change the value for this variable to the number 1. If you want to place only on the second interstitial site type,
#change it to value 2.
Run the code with:
python3 calc_int_sites.py If you want to find the interstitial sites based on the charge density method (ChargeInterstitialGenerator) instead of Voronoi method, uncomment the following two variables in the code.This approach requires the charge density CHGCAR file from VASP as input (instead of the structural POSCAR file). Ensure you generate the CHGCAR file first by performing a VASP calculation on your initial structure
generator = ChargeInterstitialGenerator(clustering_tol=1.0,min_dist=0.5)
structure = Chgcar.from_file("CHGCAR")
The periodic boundary conditions (PBC) are considered in the code within the following line:
delta = delta - np.round(delta)For example, when having two interstitial sites at [0.1, 0.1, 0.1] and [0.9, 0.9, 0.9], their distance difference with PBC (delta) should be [0.2, 0.2, 0.2] (or with minuses). This is ensured by this line, since:
delta = [0.8, 0.8, 0.8] - round([0.8, 0.8, 0.8]) = [0.8, 0.8, 0.8] - [1.0, 1.0, 1.0] = [-0.2, -0.2, -0.2]Websites for the used modules:
1. Automatically generate VASP input files
Prerequisities
- pymatgen
- Python 3.x
The following module from pymatgen.analzsis.defects finds the interstitial positions in the structure based on the charge density obtained from VASP (CHGCAR file):
from pymatgen.io.vasp.sets import MPRelaxSet, MPStaticSet from pymatgen.core import Structure from pymatgen.io.vasp import Potcar try: potcar = Potcar(["Ti", "O"]) print(potcar) except Exception as e: print(f"Error: {e}") #symbols = ["O", "Ti"] #potcar = Potcar(symbols, functional='PBE_54') user_potcar_settings = {"functional": "PBE_54"} structure = Structure.from_file("POSCAR") print(structure) vasp_input_set = MPStaticSet(structure, user_potcar_functional="PBE_54") #This generates INCAR, KPOINTS, POTCAR for single-point energy calculations
#vasp_input_set = MPRelaxSet(structure, user_potcar_functional="PBE_54") #This generates INCAR, KPOINTS, POTCAR for geometry optimization
vasp_input_set.write_input(".")
If you are using WSL, make sure there are no Zone:Identifier files, otherwise the previous command gives error. You can delete the Zone:Identifier in all subfolders using this:
cd /home/lebedmi2/DATA/potencials
find . -type f -name "*Zone.Identifier" -exec rm -f {} +