
Brief course on MACE-Interactive (MLIP-Interactive)
Introduction
Pre-trained foundation models in machine-learning interatomic potentials (MLIPs) are transforming atomistic simulations. Instead of having to design or fit a separate potential for each material, researchers can now rely on universal MLIPs (u-MLIPs) that are trained on broad, diverse datasets. These models are exposed to millions of atomic configurations across a wide variety of chemical elements, crystal structures, and bonding environments. As a result, they learn general principles of atomic interactions and can be applied to many systems without retraining.
Such models reach accuracies close to density functional theory (DFT) but are orders of magnitude faster. This enables simulations of materials with hundreds and thousands of atoms, well beyond the size limits of conventional DFT calculations.
The term foundation model is borrowed from the broader AI community, where it describes large, general-purpose models trained on massive and diverse datasets. They are called “foundation” models because they serve as a reusable base: once trained, they can be applied directly to many different problems or fine-tuned for specific tasks. In materials science, this means a single pre-trained MLIP can act as a general potential across a wide variety of chemical systems.
Several u-MLIPs are currently under active development, including:
These models are not yet finished products: they are continuously evolving, with new versions being released that improve speed, accuracy, and generality. While they are already applicable for many practical studies, users should apply them with caution: validating results whenever possible and being aware of their current limitations.
To make these models more accessible, we have developed an interactive interface that integrates MACE and the other models. It allows users to run practical calculations on multiple crystal structures at once. By simply uploading crystal structures, you can:
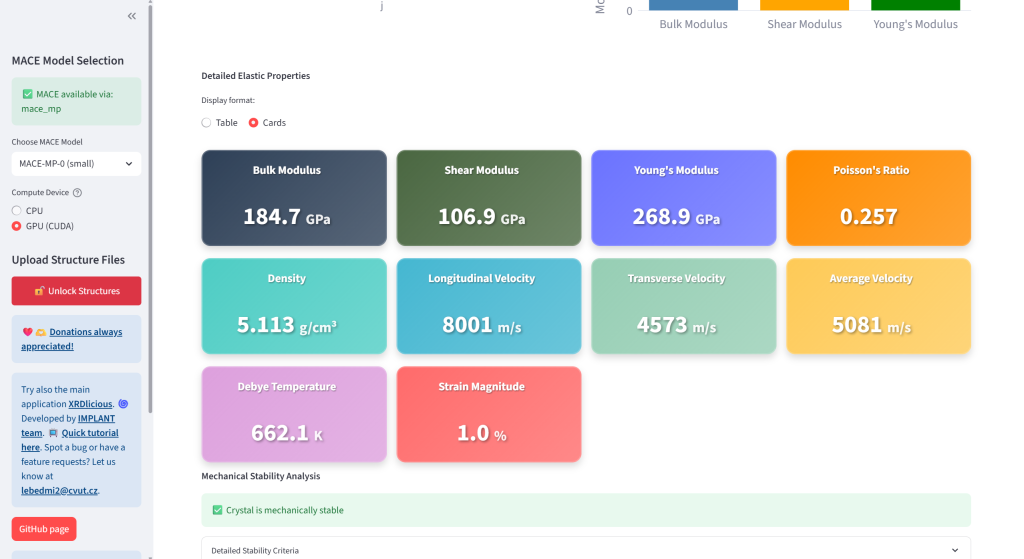
Compute single-point energies, lattice parameters, elastic properties, and phonons
Identify the most energetically favorable sites for substitutions or vacancies using a genetic algorithm
Automatically generate Python scripts that reproduce the selected calculations and execute them in a separate console, which is especially useful for automation and batch processing
In this course, we will demonstrate how to install the application and use it through hands-on examples.
.
Installation
If you do not have Linux, you have to install WSL:
uMLIP interactive (MACE gui) is working under Linux system, we are using Ubuntu system, to be able to use it under windows,
install it ubuntu in WSL (Windows subsystem linux)
in command line (cmd) write:
wsl --install -d Ubuntu
wsl.exe --install Ubuntu
if you do not have enough space in C: you can move ubuntu to D:
wsl --export Ubuntu D:\WSL\ubuntu.tar
wsl --unregister Ubuntu
wsl --import Ubuntu D:\WSL\ D:\WSL\ubuntu.tar
You will need also pythnon and another app, you can install them by:
sudo apt update
sudo apt install build-essential cmake python3-dev gfortran python3.12-venv
Now you can continue with compilation of MACE-Interactive
How to compile MACE-Interactive (MLIP-Interactive)
1) (optional but recommended) For calculations on GPU, compile CUDA:
https://developer.nvidia.com/cuda-toolkit
Do not forget to add it to the PATH, e.g.:
- In your console, open bashrc:
nano ~/.bashrc - Scroll to the end of the file
- Add the following two lines to the end of the file (modify accordingly for your version of CUDA, mine is 12.9):
export PATH=/usr/local/cuda-12.9/bin:$PATH export LD_LIBRARY_PATH=/usr/local/cuda-12.9/lib64:$LD_LIBRARY_PATH - Save the changes: press ‚ctrl + c‘, and exit the nano with ‚ctrl + x‘
- Reload the config file by writting in console:
source ~/.bashrc
2) Compile MACE-Interactive (MLIP-Interactive)
(Optional) Install Git:
sudo apt update
sudo apt install git
- Download the app code from GitHub (or download it manually without Git on the following link by clicking on ‚Code‘ and ‚Download ZIP‘, then extract the ZIP. With Git, it is automatically extracted):
git clone https://github.com/bracerino/mace-md-gui.git - Navigate to the downloaded project folder:
cd mace-md-gui/ - Create a Python virtual environment to prevent possible conflicts between packages:
python3 -m venv mace_env - Activate the Python virtual environment (before activating, make sure you are inside the mace-md-gui folder):
source mace_env/bin/activate - Install all the necessary Python packages:
pip install -r requirements.txt(with CUDA for calculations on GPU), or (pip install -r requirements-no-cuda.txt (No GPU calculations)) - Run the app (always before running it, make sure to activate its Python virtual environment (Step 4):
streamlit run app.py - To update the application, write in the main folder:
git pull
Run the MACE-Interactive (MLIP-Interactive)
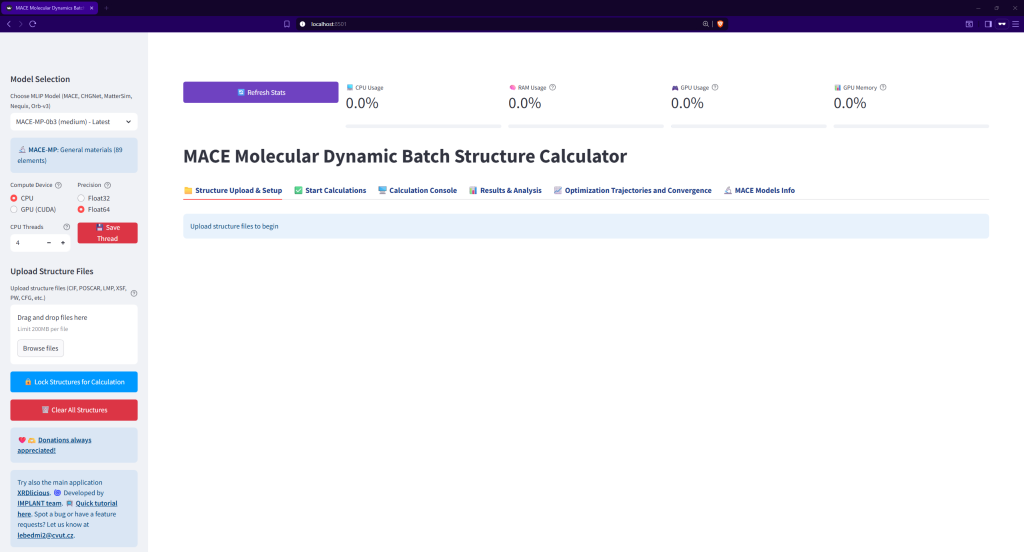
After writting:
streamlit run app.pyOpen the link for the local host (usually http://localhost:8501/) and you should see this screen:
ATOMSK – nice program for modification or creation of crystal structures
you can download the ATOMSK software https://atomsk.univ-lille.fr/dl.php (Debian/Ubuntu 64)
go to the folder where is downloaded and write:
sudo apt install ./atomsk_b0.13.1_amd64.deb
OVITO – nice program for visualization
can be used for structure visualization of several files
https://www.ovito.org/#download
https://www.ovito.org/manual/installation.html
BASH
For generation of several structures of our interest we will use simple bash script. If you are not familiar with bash
do not hesite to use chatgpt.
Task 1: Calculate elastic properties of α-Ti (hcp) and β-Ti (bcc)
1) Upload structure files
First, obtain the crystal structures of α-Ti and β-Ti. These can be provided in formats such as CIF, LMP (LAMMPS), POSCAR (VASP), or extended XYZ.
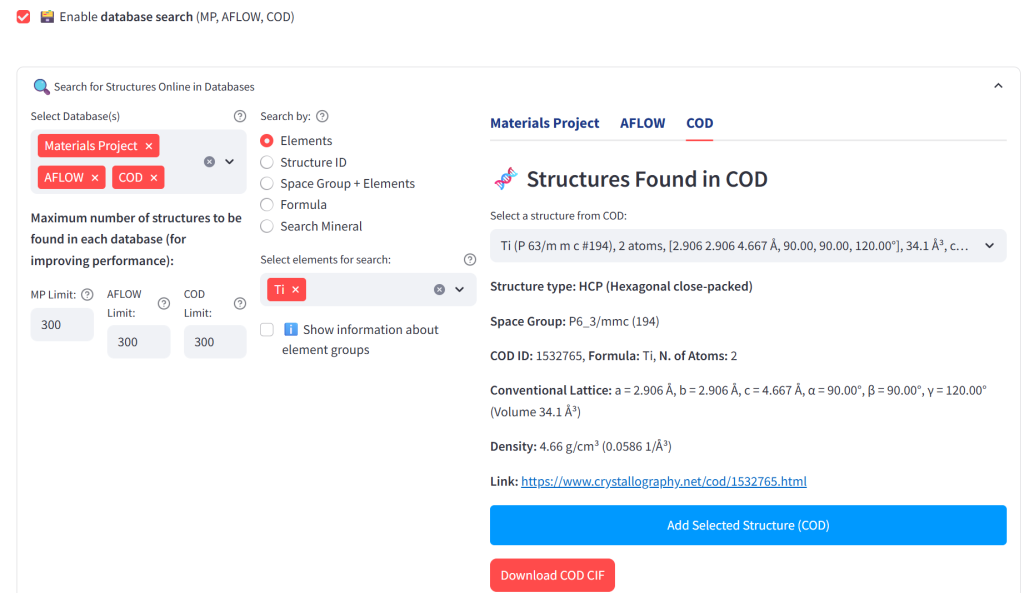
The required files are already available in the MACE-Interactive application under the course subfolder in the downloaded package. Alternatively, you can retrieve the structures from free crystallographic databases, such as the Materials Project (MP), Automatic FLOW (AFLOW), or Crystallography Open Database (COD). For convenience, you may also use our aggregator at xrdlicious.com to directly download the CIF files (see the next image).
Once you have the structure files, drag and drop both into the Upload Structure Files field in the sidebar. Then click Accept and Add Structures, followed by Lock Structures for Calculation. After that, you can preview the structures:

Task 2. Geometric optimalization of BaTiO3
============================================================================
download structure of BaTiO3 in cif format
you can use xrdlicious.com
upload it in uMLIp interactive
make geometric optimization
save the structure as o_bto.cif for using in tutorial 2.
BONUS
You can fix some coordinate a enable refinement only several of them. For this we can use VASP format of structure files – POSCAR
To create POSCAR we will use atomsk that convert cif file by following command:
atomsk bto.cif -fractional -fix all VASP
This command set all atomic position to be fixed, there is also more possibility how to fix only part of the atoms, please check the atomsk manual.
then we edit this structure file by writing nano POSCAR
The atoms that are fixed are denoted by F F F, if you would like to enable some refinement, you can change the F to T.
Task 3. Energy of system as function of Ti position in BaTiO3. =====================================================================
create or download a text file move-atom.sh in the folder where you have o_bto.cif
„““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““
# bash script that from optimezed structure file of BTO (o_bto.cif)
# creates 15 others strucutre files, with different position of Ti
for i in {1..20}; do
n=$(echo „-1 + $i/10“ | bc -l)
atomsk o_bto.cif -select Ti -shift 0 0 ${n} bto_${i}.cif
done
„“““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““
If you are using wsl and making file in windows some problem with ends of the line in text file can apper. To solve this you can use dos2unix app
you can istall it by
sudo apt update
sudo apt install dos2unix
and then you can convert a file by writing dos2unix move-atom.sh
In linux the files usually does not have right to be run. To change it you can write: chmod +x move-atom.sh
To create the structures with different position of Ti atoms you have to write
./move-atom.sh
this script generates 15 atomic structures, to have some visual insight, you can upload them to OVITO software.
You can upload all these files to uMLIP interactive and make energy calculation for all files.
Task 4. Generate random alloy of Cu-Ag system.
===============================================================================
download or create genCu-Ag.sh (nano genCu-Ag.sh)
„““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““
# bash script
atomsk –create fcc 3.614 Cu -duplicate 2 2 2 CuAg_000_1.cif # This line create pure Cu, 32 atoms in the box.
atomsk –create fcc 4.162 Ag -duplicate 2 2 2 CuAg_999_1.cif # This line create pure AG, 32 atoms in the box.
# foloving „for“ loop generate several structures, i goes for various concentration and j goes for various atom arrangement
for i in $(seq 4 4 28); do
for j in {1..5}; do
atomsk –create fcc 3.85 Cu -duplicate 2 2 2 -select random ${i} Cu -substitute Cu Ag CuAg_$((100+i))_${j}.cif
done
done
„““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““““
Upload all these files to uMLIP interactive, make geometric optimization
Task 5. Dependence of lattice parameter on vacancy concentration
============================================================================
download cubic SrTiO3 -you can use https://xrdlicious-point-defects.streamlit.app
make supercell 2x 2x 2x save it as sto24.cif
create or download a text file vac-sto.sh
“““““““““““““““““““““““““““““““““““““““““““““““““““
for i in {1..5}; do
atomsk sto24.cif -select random ${i} O -rmatom select sto_$((24-i)).cif
done
““““““““““““““““““““““““““““““““““““““““““““““““““‘
dos2unix vac-sto.sh # this line is sometimes useful if you prepare text file in windows
chmod +x vac-sto.sh # this enable to run file
./vac-sto.sh # this run the script and create 10 files
upload these files to uMLIP interactive and run geometric optimization